Application Note SMX004
Twin and multi-crystal experiments using CrysAlisPro software
Twinned or multi-crystals are often seen as challenging to work with. The prevalence of twins with both strongly and weakly diffracting domains makes it essential to have detectors high sensitivity, ultralow-noise and high dynamic range, such as HPADs or S2 CCDs, in order to best measure intensities from both domains simultaneously.
Best crystallographic practice dictates that all measurable diffraction peaks from a sample should be collected and included in refinement for the most accurate and meticulous structure determination. Rigaku Oxford Diffraction’s hardware coupled with CrysAlisPro software1 makes handling twins easier and produces better results than ever thanks to an improved data reduction algorithm.
Background
CrysAlisPro now uses simultaneous profile fitting and integration of the different twin components. Using simultaneous profile fitting, extraction of intensities gives more accurate HKLF4 intensity (single component data), and accurate HKLF5 intensity (all component data) due to better handling of overlapped reflections.
Simultaneous profile fitting also allows the use of features previously available only for single crystal samples (e.g. smart background and bad profile rejection). Combined with Ewald Explorer for indexing, CrysAlisPro makes handling twinned diffraction patterns easy and straightforward.
Classic twins
Figures 1-3 show details of twinned samples processed with CrysAlisPro and the improvement in the data by using the improved twin integration.
The triclinic sample in Figure 1 has an 80:20 split. Reprocessing with the new algorithm gives a significant improvement for both HKLF4 and HKLF 5 refinement.
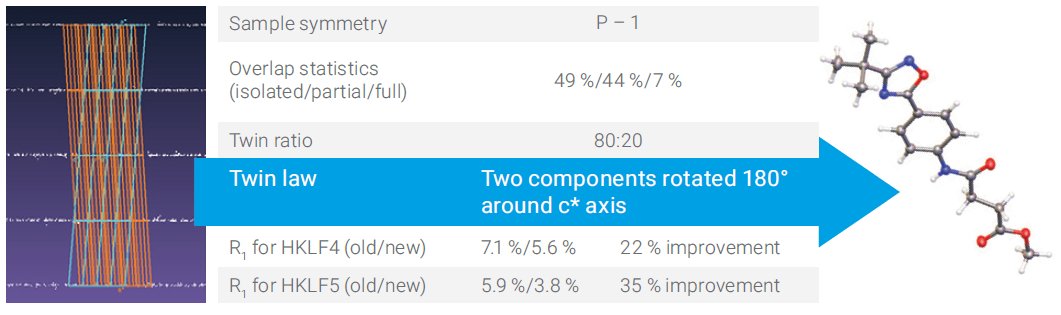 Figure 1. Triclinic sample with a major and minor twin component.
Figure 1. Triclinic sample with a major and minor twin component.
A 50:50 split monoclinic crystal is presented in Figure 2. In this example 26% of reflections involve some overlap. Here again improvements are observed when using the new algorithm.
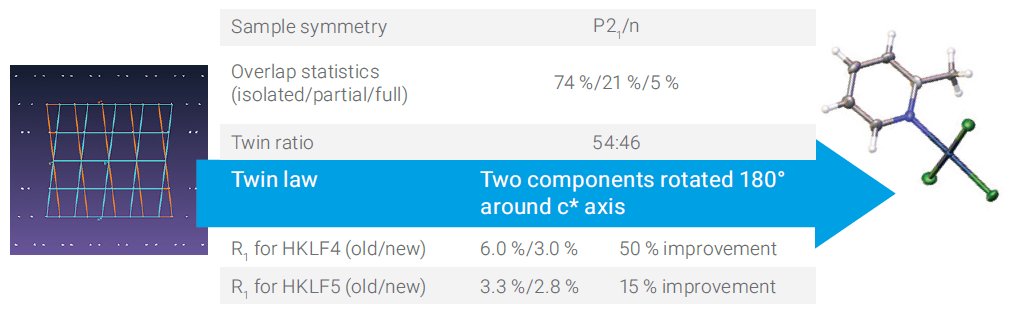 Figure 2. Monoclinic sample with roughly equal components.
Figure 2. Monoclinic sample with roughly equal components.
For this twinned protein crystal 95% of the reflections can be deconvoluted to serve as an input for structure solution and refinement.
 Figure 3. Example of a twinned protein crystal.
Figure 3. Example of a twinned protein crystal.
Data with more than two domains
In some cases, the extra information available from twin components improves data quality, so it can be very important to be able to handle these datasets. Figure 4 shows the results from a near-polycrystalline sample with four twin domains (twin ratio 45:33:15:7).
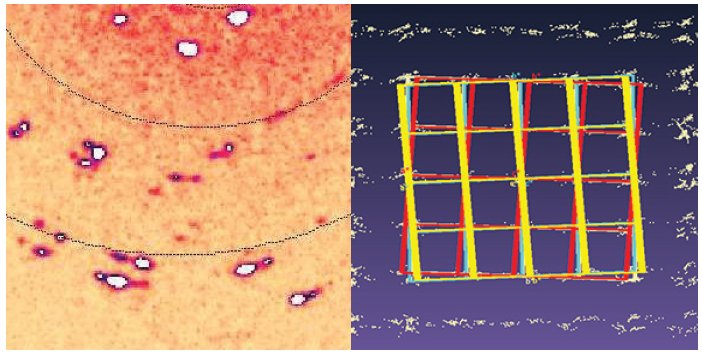 Figure 4. The diffraction image (left) clearly shows that the sample is multi-domain. By processing the four main twin components (red/orange/yellow/blue), an improvement in the refinement is possible.
Figure 4. The diffraction image (left) clearly shows that the sample is multi-domain. By processing the four main twin components (red/orange/yellow/blue), an improvement in the refinement is possible.
| Data (∞-0.8 Å) | Single crystal | 2-component refinement | 4-component refinement |
| Rint (1st component) | 7.5% | 7.3% | 7.7% |
| Rint (all components) | 7.5% | 8.1% | 9.0% |
| I/σ | 29.3 | 67.9 | 79.6 |
| R1 | 14.78% | 10.04% | 9.07% |
Multi-crystals
Two crystals of the same substance (one single and one pseudo-merohedrally twinned) were first measured individually and then together, co-mounted on the same glass fiber. The multi- crystal experiment resulted in overlap of three diffraction patterns as expected (Figure 5). The same unit cell is automatically found for each of the three domains, though CrysAlisPro does allow different unit cells for each domain. Three- component integration gave a final R1 of 5.0%. compared to 4.3% and 3.6% for the twinned and single crystals, respectively. This proof-of-concept work shows that intergrown crystals can be used for meaningful, publishable data collections when single crystals are not available.
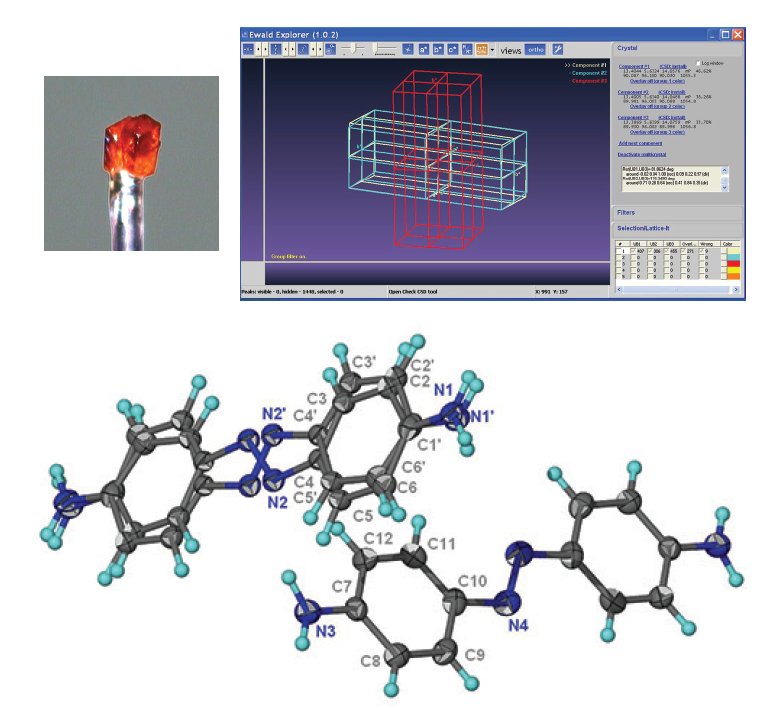 Figure 5. Co-mounted multi-crystal (left) and the lattice orientations; single (red) and twinned crystal (blue/white). Data was kindly provided by Professor Ng Seik Weng from Universiti Malaya, Kuala Lumpur2.
Figure 5. Co-mounted multi-crystal (left) and the lattice orientations; single (red) and twinned crystal (blue/white). Data was kindly provided by Professor Ng Seik Weng from Universiti Malaya, Kuala Lumpur2.
Obtaining more than one structure from a single data collection
These protein crystals always grow as aggregates of multiple crystals, and there are two polymorphs: a dimer (monoclinic C2) and a homodimer (triclinic P1), that stack along the c* axis (Figure 6). Separation is only possible via data processing, done in CrysAlisPro before solution and refinement in the CCP4 program suite4.
 Figure 6. Multi-crystal image (left), diffraction pattern (center) with the triclinic (white) and monoclinic (red) cells displayed, and the two refined structures; monoclinic C2 (left) and triclinic P1 (right). Protein data were kindly provided by Dr. Oluwatoyin Asojo from Baylor College of Medicine, Houston, Texas3.
Figure 6. Multi-crystal image (left), diffraction pattern (center) with the triclinic (white) and monoclinic (red) cells displayed, and the two refined structures; monoclinic C2 (left) and triclinic P1 (right). Protein data were kindly provided by Dr. Oluwatoyin Asojo from Baylor College of Medicine, Houston, Texas3.
 Conclusions
Conclusions
- Improved twin processing algorithms provide faster and better results with the latest version of Rigaku Oxford Diffraction CrysAlisPro.
- Multi-crystal samples can also be analyzed, and multiple structures can be obtained from a single data collection.
- It is now possible to extract structural information from more crystallographically challenging cases.
References
[1] Rigaku Oxford Diffraction, CrysAlisPro Software system, version 1.171.37.31, Rigaku Corporation, Oxford, UK, 2014.
[2] Ng Seik Weng, Chinese Journal of Structural Chemistry 2014, 33(2), pp 294-303.
[3] Asojo, O.A., et al. (In preparation).
[4] Winn, M.D., et al. Overview of the CCP4 suite and current developments. Acta. Cryst. 2011, D67, pp 235-242.
Acknowledgements
We would like to thank Prof. Seik Weng from Universiti Malaya and Dr. Asojo from Baylor College of Medicine for their contributions of data, analysis and text in this application note.
