Application Note XRD1014
Introduction
Metal-organic frameworks (MOFs), also referred to as coordination polymers, have attracted considerable interest since the early 1990s across numerous research institutions. These hybrid materials, composed of metal ions or metal-based clusters interconnected by various organic bridging ligands, are extensively studied for their unique architectures and topologies, as well as their diverse potential applications. MOFs have been extensively utilized in applications such as gas storage and separation, catalysis, drug delivery, bioimaging, magnetism, photoluminescence, sensing, optoelectronics, conductivity, and energy storage.
The crystal structures of MOFs are typically determined using single crystal X-ray or synchrotron data, or electron diffraction data. However, MOFs often crystallize as very small crystals, less than 1 μm in size, rendering standard single crystal X-ray measurements impractical, and electron diffraction methods are not always accessible. In such instances, the crystal structure of an MOF powder sample can be determined using high-resolution X-ray data collected over extended periods on floor-standing diffractometers. However, a benchtop diffractometer equipped with a high-speed 1-dimensional detector can provide high-quality data sufficient for ab-initio structure determination.
Measurement and results
The powder sample was mounted in a Si zero-background sample holder with a cavity of 10 mm x 0.2 mm and measured in reflection geometry on a Rigaku MiniFlex 600 diffractometer, equipped with a Cu-sealed tube (600 W) and a D/teX Ultra2 1-D detector. Data were recorded in the 2θ range of 5-110° with an angular step width of 0.02° and a speed of 5° min⁻¹. The total acquisition time was 22 min. Structure determination was performed using Rigaku SmartLab Studio II software. The powder pattern was indexed with N-TREOR (F20 =85.5), determining the space group I4₁/amd (no. 141), R = 1%, 85 reflections). After pattern decomposition (Rwp = 4.82, S = 3.00), the initial crystal structure model was determined using the charge-flipping method. The crystal model was refined with the Rietveld method, providing Rwp = 8.88, S = 5.21. The refined parameters included the scale factor, unit cell parameters, profile (peak shape: split pseudo-Voigt), background, atomic coordinates, and displacement parameters. The final Rietveld refinement and packing plots showing structural features of the coordination frameworks of Cu(ta)₂ are presented in Figures 1 and 2, respectively.
 Figure 1: Rietveld refinement plots of Cu(ta)₂. Red and blue lines represent observed and calculated patterns, respectively, with the difference plot shown at the bottom.
Figure 1: Rietveld refinement plots of Cu(ta)₂. Red and blue lines represent observed and calculated patterns, respectively, with the difference plot shown at the bottom.
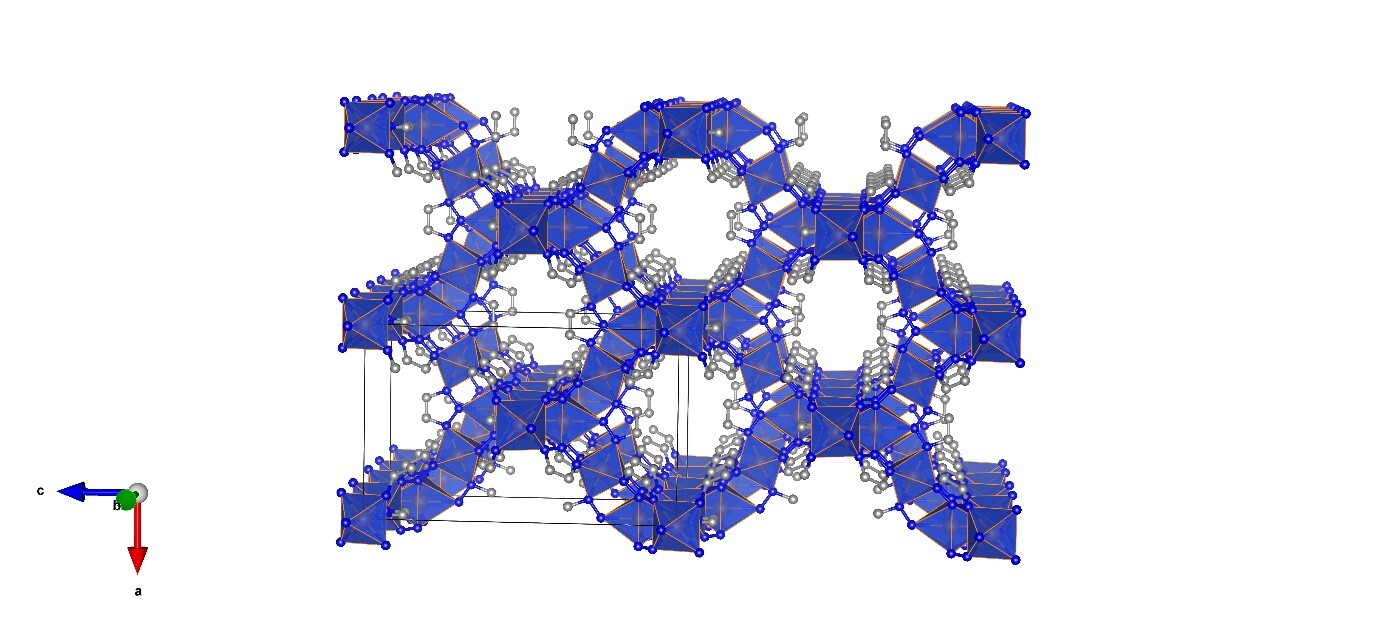 Figure 2: Packing plot showing structural features of the coordination frameworks of Cu(ta)₂.
Figure 2: Packing plot showing structural features of the coordination frameworks of Cu(ta)₂.